NMR – Rozdział 1
1. Nasze narzędzia: podstawowe techniki pomiarowe NMR i programy komputerowe wspomagające przetwarzanie i interpretację widm
Spis treści
1.1. Podstawowe techniki pomiarowe NMR.. 2
1.2. Programy do obróbki widm NMR.. 3
1.3. Komputerowe wspomaganie interpretacji widm NMR.. 9
1.3.1. Biblioteki widm NMR dostępne w Internecie. 10
1.3.3. Symulacja widm NMR.. 13
1.1. Podstawowe techniki pomiarowe NMR
Widma magnetycznego rezonansu jądrowego, a jest ich wiele rodzajów, są najskuteczniejszą metodą ustalania lub potwierdzania budowy cząsteczek związków organicznych. Najczęściej wykorzystujemy w tym celu następujące typy widm:
- standardowe (jednowymiarowe) widma 1H NMR,
- standardowe (jednowymiarowe) widma 13C NMR z pełnym odsprzęganiem sprzężeń 1H-13C oraz widma 13C DEPT,
- widma korelacyjne (dwuwymiarowe) 1H-1H COSY, 1H-13C HSQC i 1H-13C HMBC,
- widma wykorzystujące jądrowy efekt Overhausera (NOE).
Niewątpliwie najszybszym i najprostszym w wykonaniu jest jednowymiarowe widmo 1H NMR, przy czym słowo „jednowymiarowe” najczęściej się pomija. Dobrej jakości widmo 1H NMR można zarejestrować w krótkim czasie już dla 1 mg badanego związku, chociaż ilość ta zależy od wielkości cząsteczki. Takie widmo dostarcza informacji o przesunięciach chemicznych i w wielu przypadkach także o stałych sprzężenia H – H. Dla prostych związków organicznych taka informacja jest często wystarczająca, aby potwierdzić lub odrzucić proponowaną strukturę cząsteczki.
Redakcje czasopism, do których wysyłamy nasze publikacje wymagają jednak podania także widm 13C NMR opisywanych związków. Pomiar jednowymiarowego widma węglowego z pełnym odsprzęganiem sprzężeń C – H jest także prosty, ale ze względu na niską zawartość czynnego magnetycznie izotopu 13C (1,1%) pomiar wymaga znacznie większej próbki i może trwać nawet wiele godzin. W widmie zarejestrowanym tą metodą każdemu atomowi węgla (lub grupie atomów równocennych chemicznie) odpowiada pojedynczy pik. Zyskujemy w ten sposób duże skrócenie czasu rejestracji widma, ale tracimy informację o stałych sprzężenia C – H. Dla rozwiązania niektórych problemów strukturalnych jest konieczne zarejestrowanie widma 13C NMR z zachowaniem stałych sprzężenia. Służy do tego metoda nazywana w języku angielskim „gated decoupling”. Pomiar takiego widma jest dużo dłuższy od pomiaru widma z pełnym odsprzęganiem, dlatego też takie pomiary wykonuje się tylko w szczególnych przypadkach.
Do niedawna dość często wykonywanym pomiarem NMR dla jąder 13C była rejestracja widm DEPT (Distortionless Enhancement by Polarization Transfer). W najczęściej wykorzystywanym wariancie tej metody (tzw. DEPT 135°) sygnały atomów węgla grup CH3 i CH mają w widmie intensywność dodatnią, a sygnały grup CH2 – ujemną. Nie obserwuje się natomiast sygnałów czwartorzędowych atomów węgla. Należy zwrócić uwagę, że widmo DEPT 135° można sfazować też odwrotnie, tzn. sygnały atomów węgla grup CH3 i CH będą miały intensywność ujemną, sygnały atomów węgla grup CH2 – dodatnią, dlatego też trzeba mieć zidentyfikowany przynajmniej jeden sygnał atomu węgla aby pozbyć się dwuznaczności. W ten sposób można łatwo zidentyfikować atomy węgla o różnej rzędowości. Obecnie jednak znacznie częściej wykorzystuje się w tym celu dwuwymiarowe widma 1H-13C HSQC, których czas rejestracji jest dużo krótszy, a ponadto dostarczają one znacznie więcej informacji (patrz niżej).
Podczas analizy widma 1H NMR bardzo istotną informacją, którą chcemy z niego uzyskać jest ustalenie, które atomy wodoru są ze sobą sprzężone, co w praktyce oznacza, że znajdują się blisko siebie w cząsteczce. Obecnie wykorzystuje się w tym celu dwuwymiarowe widma korelacyjne 1H-1H COSY (akronim od Correlation Spectroscopy), a także – ale znacznie rzadziej – widma TOCSY (Total Correlation Spectroscopy). Liczne przykłady widm COSY oraz sposób ich interpretacji znajdują się w rozdziale 2.
Kolejną ważną informacją, którą chcielibyśmy uzyskać z pomiarów NMR jest to, które atomy wodoru są bezpośrednio związane z danymi atomami węgla. Służą do tego dwuwymiarowe widma 1H-13C HSQC (Heteronuclear Single Quantum Coherence). Z kolei dwuwymiarowe widma 1H-13C HMBC (Heteronuclear Multiple Bond Correlation) pokazują, które atomy wodoru są sprzężone z danym atomem węgla przez 2 lub 3 wiązania, a w wyjątkowych przypadkach także przez większą liczbę wiązań. Liczne przykłady widm HSQC i HMBC także znajdują się w rozdziale 2.
Jeśli pomiary NMR mają służyć do rozwiązania problemów stereochemicznych, bardzo przydatne okazują się eksperymenty, w których wykorzystuje się jądrowy efekt Overhausera (NOE – Nuclear Overhauser Effect). Obecnie rejestruje się w tym celu jedno- i dwuwymiarowe widma NOESY (Nuclear Overhauser Effect Spectroscopy). Pozwalają one zidentyfikować atomy wodoru lub równocenne magnetycznie grupy tych atomów, które znajdują się blisko siebie w cząsteczce, mimo że może dzielić je wiele wiązań. Wykorzystujemy tu efekt bezpośredniego oddziaływania na siebie jąder czynnych magnetycznie poprzez przestrzeń, a nie – jak w przypadku klasycznego sprzężenia – poprzez elektrony wiązań pomiędzy atomami. Liczne przykłady widm NOE są opisane w rozdziale 3. Niestety, dla cząsteczek o średniej wielkości efekt NOE staje się bliski zeru i w takich przypadkach pomocne jest zarejestrowanie widma ROESY (Rotating-frame nuclear Overhauser Effect Spectroscopy).
Jeśli w cząsteczce znajdują się atomy innych pierwiastków niż węgiel i wodór, które mają izotopy o niezerowym spinie jądrowym – najczęściej 19F i 31P – możemy także zarejestrować widma tych pierwiastków oraz widma korelacyjne z ich udziałem. Szczególnym przykładem jest widmo 1H-15N HMBC, które można zarejestrować jako zamiennik niezwykle czasochłonnego bezpośredniego pomiaru widma izotopu 15N. Widmom pierwiastków innych niż węgiel i wodór, a także wpływowi tych pierwiastków na wygląd widm 1H i 13C jest poświęcony rozdział 5.
Istnieje jeszcze wiele innych rodzajów pomiarów NMR, ale z punktu widzenia ustalania budowy cząsteczek związków organicznych mają one mniejsze znaczenie. Obszerną informację o podstawach teoretycznych pomiarów różnych rodzajów widm NMR można znaleźć w zamieszczonym w witrynie WWW Instytutu Chemii Organicznej PAN podręczniku pt. „Spektrometria magnetycznego rezonansu jądrowego – podstawy i metody pomiarowe” autorstwa prof. Jarosława Jaźwińskiego, a także w licznych, angielskojęzycznych podręcznikach NMR.
1.2. Programy do obróbki widm NMR
Dla większości osób zlecających wykonanie pomiarów w pracowni NMR wystarczające jest otrzymanie wyników w formie wykresów widm nadających się do interpretacji. Jednak podczas analizy widm często okazuje się, że nie wszystkie potrzebne informacje da się z tych wykresów odczytać, ponieważ personel pracowni NMR przetwarza widma w sposób rutynowy, chyba że klient precyzyjnie określi jaki jest cel wykonania pomiarów. Dlatego też chemicy zajmujący się syntezą organiczną i pokrewnymi specjalnościami, na przykład badający związki naturalne, powinni nauczyć się osobiście obrabiać widma NMR w celu uzyskania tych informacji, na których im zależy, a także przygotować wydruki widm w wersji elektronicznej do zamieszczenia w materiałach uzupełniających publikacji naukowych. Pracownie NMR mogą dostarczyć na życzenie oryginalne zestawy plików zarejestrowanych podczas pomiarów lub – jak ma to miejsce w Instytucie Chemii Organicznej PAN – udostępnić osobom upoważnionym pliki na serwerze danych. Takie zestawy plików dla wszystkich związków opisanych w tym podręczniku stanowią jego część i są dostępne przez odpowiednie łącza w spisie treści kolejnych rozdziałów (patrz Wstęp).
Osoby zainteresowane własnoręczną obróbka widm NMR mają do dyspozycji szereg programów, zarówno komercyjnych, jak i darmowych. W tym rozdziale opiszemy niektóre z nich, zwracając uwagę na ich wady i zalety. Aby w pełni wykorzystać możliwości tych programów konieczne jest zapoznanie się z instrukcjami obsługi oraz praktyczne przećwiczenie procedur przetwarzania widm różnego rodzaju. Trochę dokładniej opiszemy jedynie darmowy program SpinWorks, za pomocą którego została wykonana obróbka większości widm prezentowanych w tym opracowaniu. Więcej informacji o przetwarzaniu widm NMR z wykorzystaniem programu SpinWorks, a także komercyjnego programu MestReNova znajduje się pliku PDF dostępnym w naszej witrynie.
Zanim zaczniemy przetwarzać samodzielnie widma NMR warto wiedzieć w jakiej postaci są one zapisywane na dysku komputera. Spektrometry firmy Bruker tworzą folder dla całej serii pomiarów danej próbki. Kolejne eksperymenty są umieszczane w folderach oznaczonych liczbami. Niestety liczby te nic nie mówią o rodzaju pomiaru. Można to odczytać z pliku „pulseprogram” w danym folderze albo po prostu wczytać plik do programu do obróbki widm i zobaczyć rezultat. Przykładowe okienko eksploratora plików Windows z plikami Brukera jest pokazane na rys. 1.2.1.
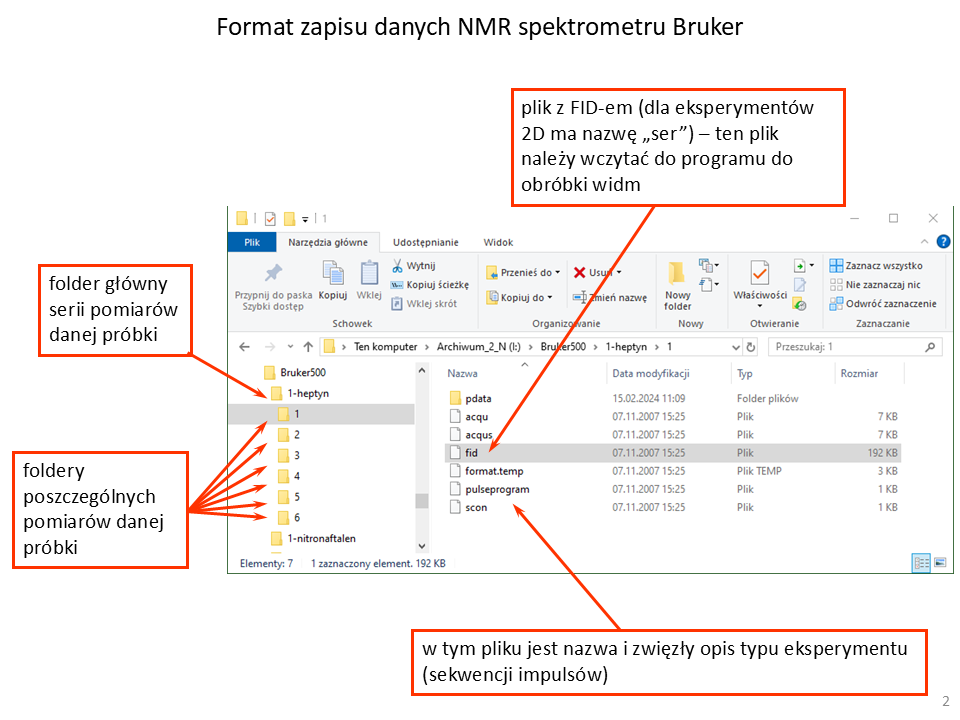
Rys. 1.2.1. Format zapisu danych pomiarów NMR spektrometrów Bruker.
Inaczej wygląda sposób zapisu wyników pomiarów widm NMR spektrometrów Agilent (dawniej Varian). Przykładowe okienko eksploratora plików Windows jest pokazane na rys. 1.2.2.
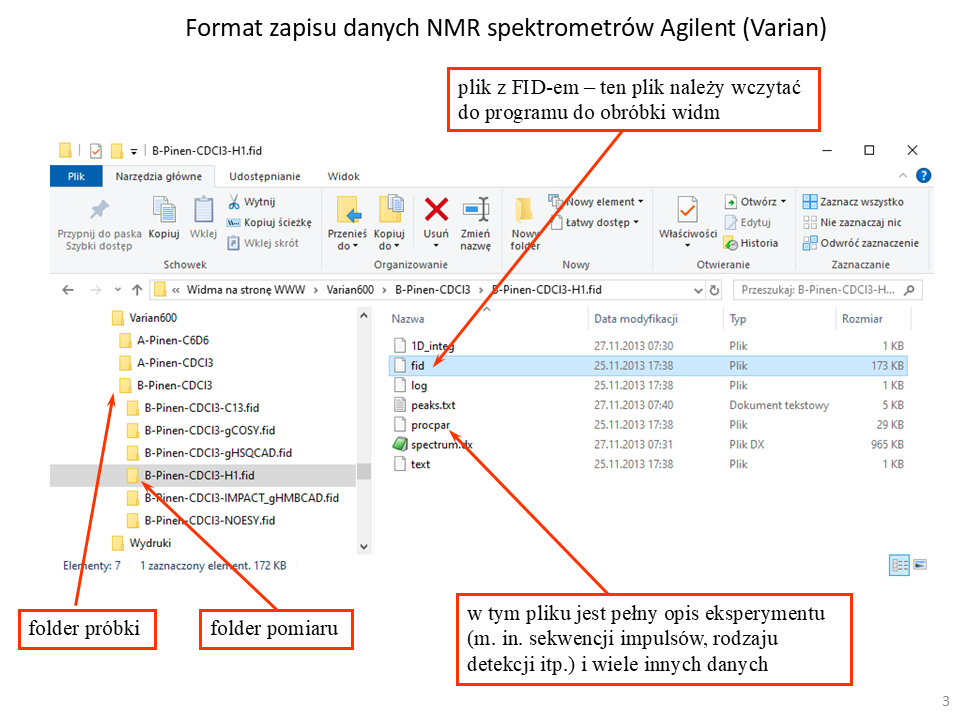
Rys. 1.2.2. Format zapisu danych pomiarów NMR spektrometrów Agilent (Varian).
Tu również mamy folder główny dla serii eksperymentów, ale każdy z podfolderów ma nazwę zakończoną znakami „fid”. Nazwa folderów zależy od operatora, ale najczęściej zawiera ona nazwę próbki, a ściślej nazwę folderu głównego oraz typ rejestrowanego widma. Bardzo ułatwia to wybór właściwego folderu do przetwarzania.
Niezależnie od typu spektrometru, do programu do obróbki widm zawsze wczytujemy plik „fid” z folderu danego eksperymentu, a w przypadku widm dwuwymiarowych rejestrowanych za pomocą spektrometrów Brukera – plik „ser”.
SpinWorks v. 4.2
Autorem programu SpinWorks jest dr Kirk Marat z Uniwersytetu Manitoba w Kanadzie, obecnie już na emeryturze. Program jest dostępny bezpłatnie i bez konieczności rejestracji pod adresem:
a także w wielu innych lokalizacjach w Internecie. Najnowsza wersja (maj 2023) ma numer 4.2.12, ale także wcześniejsze wersje są w pełni użyteczne. Procedura instalacji jest typowa dla współczesnych programów dla środowiska Windows i nie powinna stwarzać problemów. Warto jedynie zwrócić uwagę, że niektóre procedury w programie SpinWorks (np. symulacja widm) nie tolerują nazw plików i folderów ze spacjami, polskimi znakami i wieloma innymi znakami specjalnymi, dlatego dobrze jest umieszczać dane do przetwarzania w folderach bez takich znaków.
Do programu dołączona jest obszerna instrukcja, z którą warto się zapoznać, chociaż większość typowych operacji, poczynając od wczytania pliku FID, a kończąc na uzyskaniu wydruku, jest intuicyjna i po przetworzeniu kilku przykładowych widm nie stwarza już problemów. Trzeba jednak zaznaczyć, że w odróżnieniu od programów komercyjnych w programie SpinWorks nie znajdziemy automatycznych procedur ułatwiających przetwarzanie widma, a te, które są, nie działają najlepiej (np. automatyczna korekcja fazy).
Program SpinWorks umożliwia obróbkę widm jedno- i dwuwymiarowych zarejestrowanych za pomocą spektrometrów firm Bruker, Agilent (dawniej Varian) oraz Jeol, czyli praktycznie wszystkich liczących się producentów spektrometrów NMR. Trzeba jednak liczyć się z tym, że w przypadku niektórych nowszych typów widm dwuwymiarowych zarejestrowanych za pomocą spektrometrów Agilenta pewne parametry należy wpisać ręcznie. Nie dysponujemy informacjami na temat skuteczności przetwarzania widm Jeola, ponieważ nie korzystaliśmy do tej pory ze spektrometrów tej firmy. Widma wyświetlone na ekranie komputera można przenosić do programów typu Word czy PowerPoint w formacie wektorowym, czyli bez utraty jakości obrazu podczas jego skalowania.
Ważną cechą programu jest SpinWorks jest moduł symulacji widm i optymalizacji wartości przesunięć chemicznych i stałych sprzężenia tak, aby widmo symulowane jak najlepiej zgadzało się z widmem zmierzonym. Będzie o tym mowa w rozdz. 1.3.3.
Więcej informacji o przetwarzaniu widm NMR z wykorzystaniem programu SpinWorks, a także komercyjnego programu MestReNova znajduje się pliku PDF dostępnym w naszej witrynie.
TopSpin v. 4.2 (Bruker)
Program TopSpin firmy Bruker jest dołączany do spektrometrów tej firmy, ale można go też bezpłatnie pobrać ze strony producenta (https://www.bruker.com/en/products-and-solutions/mr/nmr-software/topspin.html) po zarejestrowaniu się na niej i wykorzystywać do celów niekomercyjnych. Umożliwia on przetwarzanie nie tylko widm rejestrowanych za pomocą spektrometrów Brukera, ale także Agilenta (dawniej Variana). Niestety obsługa programu jest mało intuicyjna, ponieważ wywodzi się on jeszcze z czasów sprzed ery Windows, kiedy z programów korzystało się za pomocą poleceń tekstowych. Niemniej jednak po nabraniu wprawy można za jego pomocą przetwarzać praktycznie wszystkie rodzaje widm NMR.
MestReNova (Mestre Labs)
MestReNova firm Mestre Labs jest jednym z najpopularniejszych komercyjnych programów do obróbki widm NMR. Nie zamierzmy w tym opracowaniu reklamować żadnego oprogramowania komercyjnego, dlatego zainteresowanych odsyłamy do witryny WWW programu: https://mestrelab.com/main-product/nmr.
Niektóre widma prezentowane w następnych rozdziałach tego opracowania były przetwarzane z wykorzystaniem tego programu, ponieważ program SpinWorks ma jednak ograniczone możliwości.
NMR Workbook Suite (ACD/Labs)
Firma ACD/Labs oferuje bardzo rozbudowany (i kosztowny) pakiet oprogramowania do przetwarzania widm różnych typów, a także chromatogramów gazowych i cieczowych. Jego częścią jest oprogramowanie do obróbki widm NMR o nazwie NMR Workbook Suite.
Witryna WWW: https://www.acdlabs.com/products/spectrus-platform/nmr-workbook-suite/
Jeszcze kilka lat temu była dostępna darmowa wersja programu do przetwarzania widm NMR firmy ACDLabs o nazwie ACD/NMR Processor Academic Edition. Niestety obecnie firma jej nie oferuje, ale osoby, które ten program zainstalowały, mogą nadal z niego korzystać.
iNMR
Stosunkowo mało znany komercyjny program do obróbki widm NMR, który wyewoluował z wersji darmowej. Witryna WWW programu: https://www.inmr.net.
* * *
Oprócz programów do przetwarzania widm NMR, które można zainstalować na swoim komputerze w Internecie są dostępne witryny, na których można przeprowadzić obróbkę widm on-line. Przykładem takiej witryny jest NMRium o adresie:
Potrafi ona przetwarzać jedynie widma jednowymiarowe i jej możliwości są bardzo ograniczone. Na pewno z czasem pojawi się więcej takich witryn, ale obecnie lepszym rozwiązaniem jest posiadanie odpowiedniego programu na własnym komputerze. Korzystanie z przewarzania widm on-line wymaga wysłania pliku FID do zewnętrznej witryny, co do której nie mamy pewności, co dalej z naszym widmem zrobi.
1.3. Komputerowe wspomaganie interpretacji widm NMR
Rozwój wszystkich dziedzin nauki, jaki obserwujemy od kilkudziesięciu lat, nie byłby możliwy bez szerokiego wykorzystania komputerów wyposażonych w odpowiednie oprogramowanie. Tak jest też w przypadku metod spektralnych stosowanych w chemii, w tym oczywiście spektrometrii magnetycznego rezonansu jądrowego. Współczesne spektrometry NMR są wyposażone w rozbudowane systemy komputerowe, które nie tylko sterują pomiarami, ale służą także do obróbki wyników. Zazwyczaj widma NMR są przetwarzane przez personel laboratoriów usługowych NMR do postaci wykresów nadających się do interpretacji, ale osoby zainteresowane uzyskaniem jak największej ilości informacji z wykonanych pomiarów mogą – i powinny! – przetwarzać widma osobiście. Służą do tego zarówno liczne programy komercyjne, jak i dostępne za darmo – pisaliśmy o tym w poprzednim podrozdziale. Kolejnym krokiem jest „dopasowanie” otrzymanych widm do proponowanej struktury cząsteczki badanego związku albo – w sytuacji, gdy takiej struktury nie możemy zaproponować, bo związek został np. wydzielony z materiału biologicznego – ustalenie budowy jego cząsteczki na podstawie widm. Mogą nam w tym pomóc dostępne w Internecie bazy widm NMR, przede wszystkim 1H i 13C. Tu również mamy do wyboru bazy komercyjne i – niestety dużo mniejsze – bazy darmowe.
W wielu przypadkach pomocne mogą okazać się procedury przewidywania przesunięć chemicznych na podstawie proponowanej budowy cząsteczki. Przesunięcia chemiczne można szacować na podstawie tzw. inkrementów podstawników, czyli wpływu różnych grup funkcyjnych i innych elementów cząsteczki na przesunięcia chemiczne 1H i 13C konkretnego atomu. Można to wykonać „ręcznie”, wykorzystując dostępne w podręcznikach, literaturze naukowej i w Internecie wartości inkrementów podstawników albo skorzystać z witryn internetowych, które takie obliczenia wykonają dla nas na podstawie narysowanego wzoru strukturalnego. Nawet bardziej zaawansowane programy graficzne służące do rysowania wzorów chemicznych, takie jak np. ChemDraw w wersji Professional, mają wbudowaną funkcję szacowania przesunięć chemicznych 1H i 13C na podstawie narysowanego wzoru strukturalnego. Jak przekonamy się w rozdziale 2, często tak oszacowane przesunięcia chemiczne wykazują bardzo dobrą zgodność ze zmierzonymi i umożliwiają np. rozróżnianie izomerów. Niestety zdarzają się też przewidywania całkowicie błędne, więc należy tu być ostrożnym.
Bardzo ważną rolę podczas interpretacji widm NMR odgrywają także stałe sprzężenia. Zwłaszcza stałe sprzężenia 1H-1H mają zasadnicze znaczenie podczas ustalania budowy przestrzennej badanych związków – będzie o tym mowa w rozdziale 3. W przypadku widm I-go rzędu wartości stałych sprzężenia można odczytać bezpośrednio z widma, ale w przypadku bardziej złożonych widm konieczne może być użycie programów do symulacji widm NMR. Takie programy obliczają wygląd widma NMR, czyli przesunięcia chemiczne i względne intensywności wszystkich pików w widmie z uwzględnieniem rozdzielczości spektrometru na podstawie podanych wartości przesunięć chemicznych poszczególnych atomów i stałych sprzężenia pomiędzy nimi. Dzięki temu można sprawdzić, czy wyznaczone z widma wartości tych parametrów są poprawne. Bardziej zaawansowane programy umożliwiają dopasowanie wartości przesunięć chemicznych i stałych sprzężenia tak, aby uzyskać jak najlepszą zgodność widma eksperymentalnego i obliczonego. Ma to znaczenie zwłaszcza podczas analizy widm II-go rzędu, z których nie można bezpośrednio odczytać wartości przesunięć chemicznych i stałych sprzężenia. Przykłady takich obliczeń można znaleźć w rozdziałach 2 i 3.
Podczas ustalania budowy przestrzennej cząsteczek badanych związków duże znaczenie ma przewidywanie wartości kątów dwuściennych H-C-C-H na podstawie zmierzonych wicynalnych stałych sprzężenia z wykorzystaniem różnych wariantów równania Karplusa. Liczne przykłady takiego postępowania można znaleźć w rozdziale 3. Najprostszą obecnie drogą do uzyskania informacji stereochemicznej na podstawie wicynalnej stałej sprzężenia jest obliczenie przestrzennego kształtu cząsteczki (nazywanego żargonowo geometrią cząsteczki) z wykorzystaniem odpowiednich programów komputerowych służących do modelowania molekularnego. Nawet najprostsze z nich, dostępne bezpłatnie w Internecie, które wykorzystują do obliczenia optymalnego kształtu cząsteczki mechanikę molekularną, dobrze nadają się do wyznaczania kątów dwuściennych H-C-C-H i następnie wicynalnych stałych sprzężenia. Porównując wyniki uzyskane dla różnych izomerów geometrycznych cząsteczki badanego związku można często jednoznacznie ustalić, który z nich jest właściwy.
Współczesne zaawansowane pakiety chemii kwantowej do obliczeń kształtów, energii i różnych właściwości fizykochemicznych związków organicznych mają także możliwość obliczania przesunięć chemicznych i stałych sprzężenia wszystkich atomów, których jądra są czynne magnetycznie w niezbyt dużych cząsteczkach tych związków. W ostatniej części tego rozdziału zostaną opisane takie programy i sposoby ich wykorzystania.
Zaawansowane pakiety oprogramowania NMR-owego, takie jak MestReNova oferują funkcję w pełni automatycznej lub interaktywnej interpretacji widm NMR na podstawie podanego wzoru strukturalnego analizowanego związku oraz zarejestrowanych widm. Dobre wyniki można uzyskać wówczas, gdy są dostępne zarówno widma jedno-, jak i dwuwymiarowe. Program jest w stanie wówczas zaproponować przypisania sygnałów w widmach 1H i 13C NMR odpowiednim atomom wodoru i węgla w cząsteczce. Jeśli nie zgadzamy się z propozycją podaną przez program, możemy dokonać korekty przypisań ręcznie.
I na zakończenie tego wprowadzenia trzeba wspomnieć, że burzliwie rozwijające się obecnie metody wykorzystujące sztuczną inteligencję (AI) być może już w niedalekiej przyszłości będą w stanie całkowicie zastąpić człowieka przy interpretacji nie tylko widm NMR, ale i innych widm (MS, IR, UV-VIS). Na razie jednak metody komputerowe jedynie wspomagają nas przy interpretacji widm i proponowaniu struktury cząsteczek badanych związków, dlatego też warto zapoznać się z tym, co te metody nam oferują i umieć z nich skorzystać.
1.3.1. Biblioteki widm NMR dostępne w Internecie
W Internecie są dostępne zarówno komercyjne, jak i darmowe biblioteki widm NMR. Te pierwsze są znacznie obszerniejsze i dają więcej możliwości wyszukiwania. Wadą ich jest bardzo wysoka cena, w związku z czym stać na nie tylko bogate instytucje. Biblioteki darmowe zawierają znacznie mniej widm, co nie oznacza, że nie są przydatne. Największe z nich to:
- Baza SDBS firmowana przez Japanese National Institute of Advanced Industrial Science and Technology (AIST):
Zawiera ok. 15000 widm 1H i 13C NMR, a także widma IR i MS. Dla wielu widm NMR jest podane przypisanie sygnałów w widmie do poszczególnych atomów w cząsteczce. Ma wiele możliwości wyszukiwania związków.
- Baza NMRShiftDB2 firmowana przez Uniwersytet w Kolonii:
https://nmrshiftdb.nmr.uni-koeln.de/
Zawiera ponad 68 000 widm NMR, dla wielu związków są to tylko widma 13C. Oferuje zaawansowane możliwości wyszukiwania związków, m.in. na podstawie wzoru strukturalnego. Ponadto oferuje funkcję przewidywania przesunięć chemicznych 1H i 13C na podstawie wzoru strukturalnego. Przykłady wykorzystania tej funkcji można znaleźć w rozdziale 2.
- Wiley SpectraBase:
Największa baza różnego typu widm. W wersji darmowej oferuje 10 wyszukiwań na 30 dni.
Spośród komercyjnych bibliotek widm niewątpliwie największą jest wspomniana wyżej baza Wiley SpectraBase (https://spectrabase.com/). Po wykupieniu abonamentu daje nieograniczony dostęp do widm NMR, IR, UV i MS setek tysięcy związków organicznych.
Informacje na temat widm NMR związków organicznych można też znaleźć w bazach danych SciFinder i Reaxys. Wiele instytucji naukowych w Polsce ma dostęp do tych baz. Nie znajdziemy w nich wydruków widm, a jedynie odsyłacze do oryginalnych publikacji, w których te widma były opisane.
Warto pamiętać, że zawartość Internetu zmienia się dynamicznie, w związku z czym zawsze warto sprawdzić w wyszukiwarce, czy nie pojawiły się nowe źródła danych spektralnych.
1.3.2. Szacowanie przesunięć chemicznych w widmach 1H i 13C NMR na podstawie inkrementów podstawników
Jedną z metod interpretacji widm NMR jest oszacowanie wartości przesunięć chemicznych sygnałów pochodzących od poszczególnych atomów wodoru i węgla w cząsteczce na podstawie wzoru strukturalnego tej cząsteczki. Słowo „oszacowanie” zostało użyte celowo, ponieważ nie dysponujemy obecnie metodami, które pozwoliłyby na dokładne obliczenie wartości przesunięć chemicznych atomów danej cząsteczki, nie mówiąc już o uwzględnieniu wpływu rozpuszczalnika, temperatury i stężenia próbki. Niemniej jednak istnieją metody, które pozwalają dokonać takiego oszacowania dla widm większości typowych związków organicznych rejestrowanych w standardowych warunkach, czyli w temperaturze pokojowej i w niepolarnym rozpuszczalniku, takim jak CDCl3. Określenie „typowych” oznacza związki, w których cząsteczkach nie występują efekty steryczne, takie jak zahamowana rotacja czy inwersja pierścienia (patrz rozdz. 4).
Najczęściej stosowana metoda wykorzystuje tzw. inkrementy podstawników. Opiera się ona na założeniu, że każdy podstawnik związany z danym atomem węgla wywiera określony wpływ na przesunięcie chemiczne tego atomu oraz związanych z nim atomów wodoru, a także na przesunięcia atomów odległych o 2 lub 3 wiązania. Drugim podstawowym założeniem jest sumowanie się wpływów poszczególnych podstawników. Te wpływy są określane właśnie mianem inkrementów. Przy takich założeniach wartość przesunięcia chemicznego danego atomu jest sumą pewnej wartości „bazowej” oraz inkrementów podstawników. Wartości inkrementów podstawników zależą od hybrydyzacji atomu węgla, z którym są związane. Dlatego też mamy zestawy inkrementów dla związków nasyconych, alkenów, alkinów i związków aromatycznych. W przypadku związków nienasyconych i aromatycznych dodatkową komplikacją jest wzajemne oddziaływanie podstawników w wyniku sprzężenia. Dlatego też stosuje się poprawki dla poszczególnych par podstawników w określonych położeniach w cząsteczce. Na przykład, jeśli chcemy obliczyć przesunięcia chemiczne atomów węgla w pierścieniu benzenowym musimy brać pod uwagę nie tylko inkrementy związanych z nim podstawników, ale też ich wzajemne położenie: orto, meta lub para względem siebie.
Wartości inkrementów podstawników zostały obliczone na podstawie danych uzyskanych w wyniku pomiarów widm NMR tysięcy związków. W podręcznikach NMR oraz w Internecie można znaleźć wiele takich zestawów i wykorzystać je do swoich celów. Tablice inkrementów można znaleźć m. in. w podręczniku R.M. Silverstein, F.X. Webster, D.J. Kiemle „Spektroskopowe metody identyfikacji związków organicznych” PWN 2007. Co ciekawe, z Internetu zniknęło ostatnio kilka stron z takimi danymi. Tablice inkrementów dla benzenu oraz związków alifatycznych można jeszcze znaleźć pod adresem:
https://www.chem.ucla.edu/~bacher/General/30BL/NMR/
ale nie wiadomo jak długo będą jeszcze dostępne. Podany wyżej adres kieruje do folderu z plikami, z których te z rozszerzeniem .html kierują do odpowiednich tablic inkrementów. Kopie tych tablic można znaleźć w pliku PDF dołączonym do tego opracowania pod adresem „Inkrementy.pdf”.
Brak tablic inkrementów w Internecie wynika najprawdopodobniej z tego, że są dostępne darmowe witryny, które wykonują obliczenia przesunięć chemicznych z wykorzystaniem inkrementów podstawników dla dowolnych związków organicznych na podstawie ich wzoru strukturalnego. W tym opracowaniu korzystaliśmy z wspomnianej już w poprzednim podrozdziale witryny NMRShiftDB2 firmowanej przez Uniwersytet w Kolonii:
https://nmrshiftdb.nmr.uni-koeln.de/
Jest to nie tylko baza danych widm NMR, ale także narzędzie do przewidywania przesunięć chemicznych 1H i 13C związków organicznych na podstawie ich wzorów strukturalnych. Wykorzystywana jest w tym celu metoda inkrementów podstawników. W przypadku jednak, gdy widmo 1H i/lub 13C związku o podanej strukturze znajduje się w bazie danych, wyświetlane są dane doświadczalne zamiast przewidywanych. Dlatego też nie należy się dziwić, że dla popularnych związków trafność „przewidywania” jest znakomita. Dla związków nieznanych procedura predykcyjna ocenia także na ile wiarygodne są proponowane wyniki i zaznacza odpowiednio te wartości, dla których można spodziewać się dużych błędów.
Podobna funkcjonalność, chociaż bez bazy widm, ale za to z wieloma innymi funkcjami, mają dwie witryny nmrdb.org i cheminfo.org o adresach:
https://www.nmrdb.org/13c/index.shtml?v=v2.138.0#
https://www.cheminfo.org/Spectra/NMR/Predictions/13C_Prediction/index.html
Są one bardzo podobne do siebie, a moduły do przewidywania przesunięć chemicznych są identyczne. Za ich pomocą można przewidywać przesunięcia chemiczne 1H i 13C związków organicznych na podstawie wzoru strukturalnego. Inna wersja tej witryny jest dostępna pod adresem:
https://www.nmrium.org/predict
Te witryny oferują także inne funkcje, w tym obróbkę widm jednowymiarowych, symulację widm, a także materiały szkoleniowe. Warto się z nimi zapoznać.
Funkcję przewidywania przesunięć chemicznych mają także komercyjne pakiety, takie jak Spectrus firmy ACD/Labs oraz MestReNova firmy Mestre Labs. Korzystają one z różnych algorytmów, a dokładność proponowanych wartości przesunięć chemicznych jest na ogół bardzo dobra.
I na koniec trzeba wspomnieć, że funkcję przewidywania przesunięć chemicznych 1H i 13C ma także popularny program do rysowania wzorów chemicznych ChemDraw w wersji Professional. Nasze doświadczenia wskazują, że dla prostych związków dokładność przewidywania przesunięć chemicznych jest dobra, ale niestety często zdarzają się przewidywania bardzo odbiegające od wartości eksperymentalnych. Dlatego też, korzystając z takich programów należy zachować dużą dozę krytycyzmu. Wiele przykładów wykorzystania tego programu znajduje się w rozdziale 2.
1.3.3. Symulacja widm NMR
Jeśli celem naszej pracy było wyznaczenie wartości przesunięć chemicznych i stałych sprzężenia w widmie 1H NMR, możemy potwierdzić ich poprawność wykonując symulację widma. Służą do tego programy komputerowe, które na podstawie wyznaczonych przez nas parametrów oraz częstotliwości spektrometru i zadanej szerokości połówkowej sygnałów obliczają teoretyczny wygląd widma. Sprawa jest bardzo prosta w przypadku widm I-go rzędu, czyli takich, w których różnice wartości przesunięć chemicznych są znacznie większe od wartości stałych sprzężenia pomiędzy atomami wodoru. Z takich widm można bezpośrednio odczytać wartości przesunięć chemicznych i stałych sprzężenia, więc symulacja ma na celu jedynie potwierdzenie, czy nie popełniliśmy błędów rachunkowych.
Dużo bardziej złożona sytuacja jest w przypadku widm II-go rzędu, w których wartości stałych sprzężenia są porównywalne z różnicami wartości przesunięć chemicznych. Z takiego widma możemy jedynie oszacować przybliżone wartości parametrów spektralnych, a często i to jest niemożliwe i musimy posłużyć się wartościami przesunięć chemicznych oszacowanych za pomocą odpowiednich programów komputerowych (patrz rozdz. 1.3.2 i 1.3.5) i typowymi wartościami stałych sprzężenia dla założonej struktury cząsteczki. Po wprowadzeniu tych danych do programu symulacyjnego przy pewnej dozie szczęścia otrzymamy widmo zbliżone wyglądem do widma doświadczalnego. Dla uzyskania lepszej zgodności można modyfikować wybrane parametry, ale wymaga to cierpliwości i dużej praktyki. Bardziej zaawansowane programy do symulacji widm umożliwiają przypisanie sygnałów w widmie symulowanym odpowiednim sygnałom w widmie doświadczalnym, a następnie uściślają wprowadzone parametry d i J tak, aby widmo obliczone w jak największym stopniu zgadzało się z widmem zmierzonym. Właściwy dobór danych wejściowych oraz przypisanie sygnałom w widmie symulowanym sygnałów w widmie zmierzonym wymaga dużej praktyki, dlatego też jest to raczej domena specjalistów w dziedzinie NMR.
Modułami do prostej symulacji widm NMR dysponuje wiele programów zarówno darmowych, jak i komercyjnych. Bardzo dobrze nadaje się do tego np. program SpinWorks. Jako jedyny zawiera on także procedury uściślania parametrów spektralnych. Dokładny opis korzystania z tych procedur można znaleźć w instrukcji do programu. Liczne przykłady symulacji widm 1H NMR za pomocą programu SpinWorks, w tym także przykłady symulacji widm II-go rzędu z uściślaniem parametrów d i J można znaleźć w rozdziałach 2 i 3.
Moduł symulacji widm 1H NMR w trybie online znajdziemy także w witrynach nmrdb.org i cheminfo.org:
https://www.nmrdb.org/simulator/index.shtml?v=v2.121.2
https://www.cheminfo.org/Spectra/NMR/Tools/Simulate_spin_system/index.html
Nie oferują one jednak uściślania parametrów widma, a także – co jest poważnym utrudnieniem – nie pozwalają na podanie liczby równocennych magnetycznie atomów wodoru.
Także komercyjne pakiety programów ACD/Labs i MestReNova (patrz rozdz. 1.1) oferują moduły symulacji widm. Wybór jest więc szeroki.
1.3.4. Modelowanie molekularne i obliczenia stałych sprzężenia z wykorzystaniem równania Karplusa i jego modyfikacji
W dzisiejszych czasach łatwej dostępności widm dwuwymiarowych rola stałych sprzężenia jako ważnej informacji strukturalnej bardzo zmalała, ale ciągle istnieją liczne problemy strukturalne, a zwłaszcza stereochemiczne, do rozwiązania których znajomość wartości stałych sprzężenia jest niezbędna. Liczne przykłady takiego wykorzystania stałych sprzężenia zostały opisane w rozdziale 3. Często spotykanym przypadkiem jest wykorzystanie tzw. wicynalnej stałej sprzężenia do oszacowania wartości kąta dwuściennego utworzonego przez ugrupowanie H–C–C–H na podstawie równania Karplusa. Szczegółowe informacje na ten temat znajdują się w rozdziale 3.3.
Jeszcze częściej równanie Karplusa jest wykorzystywane w drugą stronę, tzn. na podstawie wartości kąta dwuściennego H–C–C–H w modelu cząsteczki obliczamy wicynalną stałą sprzężenia i porównujemy z wartością wyznaczoną ze zmierzonego widma. Kluczowym etapem tej procedury jest stworzenie możliwie dokładnego modelu cząsteczki. Obecnie takie modele tworzy się za pomocą odpowiednich programów komputerowych. Te programy wykorzystują różne metody modelowania molekularnego, poczynając od najprostszej metody, którą jest mechanika molekularna do najbardziej zaawansowanych metod chemii kwantowej. O tych ostatnich powiemy więcej w następnym rozdziale, tu natomiast zajmiemy się programami, które do tworzenia przestrzennych modeli cząsteczek wykorzystują mechanikę molekularną.
Nie wdając się w szczegóły, w tej metodzie cząsteczkę traktuje się jako zbiór punktowych atomów o danej masie i typie hybrydyzacji połączonych wiązaniami, które charakteryzują się określonymi parametrami mechanicznymi, takimi jak energia wymagana do rozciągnięcia wiązania, wygięcia go lub skręcenia o określony kąt. Te parametry zostały dobrane empirycznie na podstawie informacji o budowie przestrzennej setek i tysięcy związków organicznych. Ta metoda, mimo swojej prostoty, bardzo dobrze nadaje się do modelowania cząsteczek prostych związków organicznych w fazie gazowej. Przejście z fazy gazowej do rozcieńczonego roztworu w niepolarnym rozpuszczalniku nie zmienia istotnie kształtu cząsteczki, więc przyjmuje się, że mechanika molekularna dobrze odtwarza kształty cząsteczek np. w deuterochloroformie, czyli rozpuszczalniku najczęściej wykorzystywanym w pomiarach NMR. Niewątpliwą zaletą mechaniki molekularnej jest szybkość działania – obecne komputery osobiste wykonują takie obliczenia praktycznie w czasie rzeczywistym dla cząsteczek o średniej wielkości.
Jak zwykle, także i w przypadku programów do modelowania molekularnego dysponujemy zarówno programami darmowymi, jak i komercyjnymi. Pierwszym programem do obliczeń metodami mechaniki molekularnej, który pojawił się na rynku na początku lat 90. XX wieku był PCModel. Program ten nie jest już rozwijany, ale jego wersja 10 jest dostępna za darmo w Internecie, np. na stronie:
http://www.serenasoft.com/license.html
Budowanie „od zera” modelu cząsteczki w tym programie jest dość uciążliwe i mało intuicyjne. Dlatego też lepiej skorzystać z innych programów służących do tego celu, zapisać utworzony model na dysku w jednym z formatów, które są rozpoznawane przez PCModel, wczytać ten plik do programu i wykonać optymalizację geometrii cząsteczki. Bardzo użyteczną cechą programu PCModel jest wyświetlanie na ekranie wartości wicynalnych stałych sprzężenia obliczonych na podstawie modyfikowanego równania Karplusa. Przykład wykorzystanie programu PCModel jest opisany w rozdz. 3.3.3.
Drugim darmowym programem do modelowania molekularnego za pomocą mechaniki molekularnej jest Avogadro. Jest on dostępny pod adresem:
Jest to dość rozbudowany program służący do budowania modeli cząsteczek, optymalizacji ich geometrii metodami mechaniki molekularnej oraz wizualizacji wyników obliczeń geometrii cząsteczek wykonanych innymi programami. Z naszego punktu widzenia najważniejsza jest możliwość stosunkowo prostego budowania modelu cząsteczki i optymalizacji jej geometrii praktycznie w czasie rzeczywistym. Dla zoptymalizowanej cząsteczki możemy odczytać wartości odpowiednich kątów dwuściennych, a następnie, korzystając z równania Karplusa, obliczyć odpowiadające im stałe sprzężenia. Obliczania wartości stałych sprzężenia nie trzeba wykonywać „ręcznie”, ponieważ w Internecie są dostępne witryny, które zrobią to za nas. Jedna z takich witryn jest dostępna pod adresem:
http://www.stenutz.eu/conf/haasnoot.php
Funkcję budowania „od podstaw” modelu cząsteczki, a następnie optymalizację jej kształtu oferują też liczne programy komercyjne. Z punktu widzenia użytkownika chyba najprostszym w użyciu jest program Spartan, obecnie w wersji 24. Informacje o tym programie można znaleźć w witrynie producenta:
https://store.wavefun.com/default.asp
Popularnym, komercyjnym pakietem oprogramowania do obliczeń metodami mechaniki molekularnej i metodami chemii kwantowej jest platforma Maestro firmy Schrödinger:
https://www.schrodinger.com/platform/products/maestro/
Profesjonaliści w dziedzinie obliczeń metodami chemii kwantowej chętnie korzystają z programu Gaussian, obecnie w wersji 16. W Polsce jest on dostępny dla pracowników naukowych i studentów w wielu centrach superkomputerowych za pośrednictwem systemu PLGrid (https://plgrid.pl/), Wystarczy założyć tam konto. Oczywiście istnieją też wersje Gaussiana, które można zainstalować na komputerze osobistym pracującym pod kontrolą Windows lub stacji roboczej pod kontrolą systemu Linux, ale ich ceny są wysokie. Należy też pamiętać, że Gaussian nie ma interfejsu graficznego, w związku z czym przygotowanie danych do obliczeń wymaga stworzenia pliku tekstowego z odpowiednimi danymi i komendami. To samo dotyczy obrazowania wyników. Można też skorzystać z programu GaussView dla Windows (obecnie w wersji 6), również oferowanego przez firmę Gaussian, za pomocą którego można łatwo zbudować model cząsteczki i przygotować plik wsadowy do obliczeń. Dotyczy to jednak tylko bardzo ograniczonej liczby typów obliczeń, dlatego też najczęściej po stworzeniu pliku za pomocą programu GaussView, trzeba go modyfikować w edytorze tekstowym. Co ważniejsze GaussView pozwala na wyświetlenie wszystkich istotnych wyników wykonanych obliczeń, dlatego też warto go posiadać. Informacje o programach Gaussian i GaussView są dostępne na stronie:
1.3.5. Obliczanie przesunięć chemicznych i stałych sprzężenia z wykorzystaniem metod chemii kwantowej.
Rozwój teorii obliczeń kwantowo-chemicznych oraz gwałtowny wzrost wydajności systemów komputerowych w ostatnich latach umożliwił obliczanie wartości przesunięć chemicznych (ściślej: stałych ekranowania) oraz stałych sprzężenia na podstawie modelu cząsteczki. Obecnie na rynku są dostępne komercyjne pakiety, które można wykorzystać w tym celu. Są to, między innymi. wspomniane już w poprzednim podrozdziale pakiety Gaussian i Spartan, a także moduł Jaguar pakietu programów firmy Schrödinger:
https://www.schrodinger.com/platform/products/jaguar-spectroscopy/
Osoby, które nie miały jeszcze do czynienia z obliczaniem przesunięć chemicznych i stałych sprzężenia metodami chemii kwantowej mogą zacząć od zapoznania się z artykułem pt. „A guide to small-molecule structure assignment through computation of (1H and 13C) NMR chemical shifts” opublikowanym w czasopiśmie Nature Protocols (Nature Protocols, 2014, 9, 643, DOI: 10.1038/nprot.2014.042; do tego artykułu zostało opublikowane uzupełnienie: Nature Protocols, 2020, 15, 2277, DOI: 10.1038/s41596-020-0293-9). Dodajmy od razu, że czasy obliczeń podane w artykule są już dawno nieaktualne – obecnie takie obliczenia wykonuje się kilkakrotnie szybciej. Warto też odwiedzić witrynę internetową http://cheshirenmr.info/, na której można znaleźć dużo informacji o obliczeniach kwantowo-chemicznych w dziedzinie NMR wraz z przykładami. Na pewno warto zapoznać się z metodyką obliczania widm NMR z wykorzystaniem metod chemii kwantowej, ponieważ zyskują one coraz bardziej na znaczeniu i stają się wręcz rutynowym narzędziem wspomagającym interpretację widm. Przykłady zastosowania tych obliczeń można znaleźć w kolejnych rozdziałach tego podręcznika.
W ogólnym przypadku procedura obliczeniowa składa się z następujących kroków:
- Wykonanie obliczeń za pomocą mechaniki molekularnej do wygenerowania biblioteki konformerów.
- Wykonanie obliczeń wykorzystujących teorię funkcjonałów gęstości (DFT) do określenia optymalnej geometrii, energii swobodnej Gibbsa i stałych ekranowania dla każdego konformeru w zadanym przedziale energii.
- Przeliczenie stałych ekranowania na wartości przesunięć chemicznych z wykorzystaniem stałych ekranowania obliczonych tą samą metodą dla związku wzorcowego, czyli najczęściej tetrametylosilanu (TMS).
- Obliczenie przesunięć chemicznych atomów węgla i wodoru jako średnich ważonych przesunięć chemicznych poszczególnych konformerów z wykorzystaniem rozkładu Boltzmanna.
- Porównanie zmierzonych i obliczonych wartości przesunięć chemicznych na wykresie – powinny one utworzyć linię prostą.
Dodajmy jeszcze, że obliczenia są wykonywane dla „zamrożonej” cząsteczki. Na przykład, dla poszczególnych atomów wodoru grupy metylowej otrzymamy, w zależności od symetrii cząsteczki, dwa lub trzy różne wyniki, z których należy obliczyć średnią arytmetyczną, aby otrzymać poprawną wartość przesunięcia chemicznego grupy CH3. To samo dotyczy atomów wodoru w grupach CH2 z wyjątkiem cząsteczek, w których ze względów geometrycznych atomy wodoru grupy metylenowej są nierównocenne magnetycznie (patrz Rozdział 3).
Znaczącym uproszczeniem jest wykonywanie obliczeń dla cząsteczek w fazie gazowej. Zasadniczo można – przynajmniej w programie Gaussian – wykonać obliczenia, stosując jeden z modeli uwzględniających wpływ rozpuszczalnika zarówno na kształt cząsteczki, jak i jej parametry termodynamiczne, ale w praktyce nakład pracy nie jest wart uzyskanych rezultatów. Dlatego też wszystkie obliczenia opisane w tym rozdziale, a także w dalszej części podręcznika były wykonane w fazie gazowej.
Jako przykład zastosowania tej procedury pokażemy tu obliczenia wartości przesunięć chemicznych 1H i 13C cząsteczek octanu winylu i akrylanu metylu za pomocą programów Gaussian i Spartan. Pełna analiza widm 1H i 13C tych związków jest opisana w rozdziale 2.1.2.
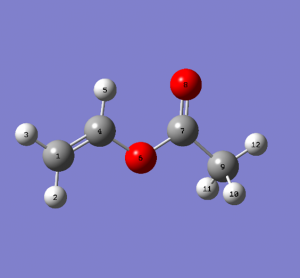
Zacznijmy od octanu winylu i programu Gaussian. Analiza konformacyjna wskazuje, że jeden z konformerów (pokazany na rysunku poniżej) jest zdecydowanie najtrwalszy, co znacznie upraszcza dalsze postępowanie. Optymalizację geometrii jego cząsteczki wykonano metodą B3LYP/6-31G(d), a następnie dla tej geometrii przeprowadzono obliczenia stałych ekranowania jąder 1H i 13C metodą B3PW91/6-311+G(2d,p). Wyniki obliczeń pokazane są w Tabeli 1.3.1.
Tabela 1.3.1. Wyniki obliczenia przesunięć chemicznych 1H i 13C w cząsteczce octanu winylu za pomocą programu Gaussian.
Stałe ekranowania obliczone dla wzorca (TMS): H: 31,8821 C: 182,4656
| Atom | Stała ekranowania | d obl. | d obl. uśrednione | d eksp. |
| 2-H | 26,7634 | 5,1187 | 5,12 | 4,88 |
| 3-H | 27,1723 | 4,7098 | 4,71 | 4,57 |
| 5-H | 23,7364 | 8,1457 | 8,15 | 7,27 |
| 10-H | 29,594 | 2,2881 | 2,18 | 2,14 |
| 11-H | 29,594 | 2,2881 | ||
| 12-H | 29,9247 | 1,9574 | ||
| 1-C | 87,2623 | 95,2033 | 95,2 | 97,5 |
| 4-C | 36,9697 | 145,4959 | 145,5 | 141,1 |
| 7-C | 12,8835 | 169,5821 | 169,6 | 167,9 |
| 9-C | 163,9605 | 18,5051 | 18,5 | 20,6 |
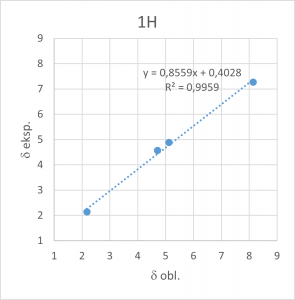
Wartości przesunięć chemicznych oblicza się, odejmując obliczoną wartość stałej ekranowania danego atomu od wartości stałej ekranowania tetrametylosilanu (TMS) obliczonej tą samą metodą. Kolejnym krokiem jest obliczenie średniej arytmetycznej wartości przesunięć chemicznych trzech atomów wodoru grupy metylowej, ponieważ są one równocenne magnetycznie za względu na szybki obrót tej grupy wokół wiązania C–C. Porównanie wartości zmierzonych i obliczonych wartości przesunięć chemicznych ujawnia znaczące różnice, ale dokładniejsza analiza wskazuje, że błąd jest systematyczny i ławo go skorygować. W tym celu należy narysować wykresy zależności zmierzonych przesunięć chemicznych od obliczonych (lub odwrotnie). Są one pokazane na rys. 1.3.1.
 |
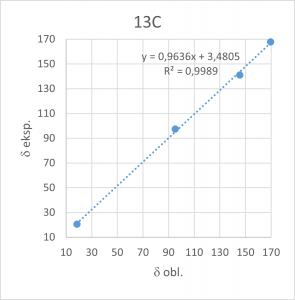 |
Rys. 1.3.1. Wykresy zależności zmierzonych wartości przesunięć chemicznych 1H i 13C od wartości obliczonych dla octanu winylu.
Jak widać, współczynniki korelacji są dla obu wykresów bardzo wysokie. Jest to wystarczające potwierdzenie poprawności interpretacji widm 1H i 13C i równocześnie tego, że pomiary widm zostały rzeczywiście wykonane dla octanu winylu.
Trochę trudniejsze jest obliczenie przesunięć chemicznych 1H i 13C w przypadku akrylanu metylu. Analiza konformacyjna wskazuje, że ta cząsteczka może występować w formie dwóch konformerów niewiele różniących się energią. Ich zoptymalizowane kształty są pokazane w Tabeli 1.3.2. W tej tabeli są też zestawione wyniki obliczeń wartości przesunięć chemicznych jąder 1H i 13C dla obu konformerów. Jak widać, w przypadku tego związku różnice nie są duże, ale jednak znaczące, zwłaszcza dla atomu wodoru H-2. Dlatego też zostały dodatkowo wykonane obliczenia parametrów termodynamicznych obu konformerów. W ich wyniku stwierdzono, że konformer 2 jest o 0,67 kcal/mol trwalszy, czyli jego swobodna energia Gibbsa jest o tyle niższa od energii konformeru 1. W ogólnym przypadku populacje poszczególnych konformerów oblicza się z równania Boltzmanna:
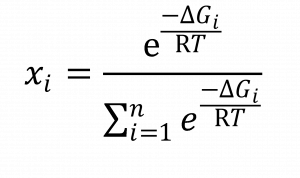
W tym równaniu xi to ułamek molowy konformeru i, a DGi to różnica swobodnej energii Gibbsa konformeru i i konformeru o najniższej energii. W naszym przypadku ułamek molowy konformeru 1 jest równy 0,24, a konformeru 2 – 0,76. Aby więc obliczyć średnie ważone wartości przesunięć chemicznych wartość dla konformeru 1 mnożymy przez 0,24 i dodajemy do wartości dla konformeru 2 pomnożonej przez 0,76. Wyniki znajdują się w Tabeli 1.3.2.
Tabela 1.3.2. Wyniki obliczenia przesunięć chemicznych 1H i 13C w cząsteczce akrylanu metylu za pomocą programu Gaussian.
 |
 |
| Geometria 1 | Geometria 2 | |||||||||
| Atom | Stała ekranowania | d obl. | d obl. uśrednione | Stała ekranowania | d obl. | d obl. uśrednione | Średnia ważona | d eksp. | ||
| 2-H | 25,1117 | 6,7704 | 6,77 | 24,774 | 7,1081 | 7,11 | 7,03 | 6,41 | ||
| 5-H | 25,3749 | 6,5072 | 6,51 | 25,2693 | 6,6128 | 6,61 | 6,59 | 6,13 | ||
| 3-H | 25,6258 | 6,2563 | 6,26 | 25,6988 | 6,1833 | 6,18 | 6,20 | 5,83 | ||
| 11-H | 27,9549 | 3,9272 | 3,82 | 27,9411 | 3,941 | 3,82 | 3,82 | 3,76 | ||
| 12-H | 27,9554 | 3,9267 | 27,9413 | 3,9408 | ||||||
| 10-H | 28,2747 | 3,6074 | 28,2953 | 3,5868 | ||||||
| 1-C | 49,4334 | 133,0322 | 133,0 | 46,8974 | 135,5682 | 135,6 | 135,0 | 130,7 | ||
| 4-C | 48,5687 | 133,8969 | 133,9 | 50,9639 | 131,5017 | 131,5 | 132,1 | 128,1 | ||
| 6-C | 14,3466 | 168,119 | 168,1 | 13,0531 | 169,4125 | 169,4 | 169,1 | 166,6 | ||
| 9-C | 132,2842 | 50,1814 | 50,2 | 132,1822 | 50,2834 | 50,3 | 50,3 | 51,5 | ||
Podobnie jak w opisanym wyżej przypadku octanu winylu, kolejnym krokiem jest sporządzenie wykresów zależności eksperymentalnych wartości przesunięć chemicznych od wartości obliczonych. Wykresy są pokazane na rys. 1.3.2.
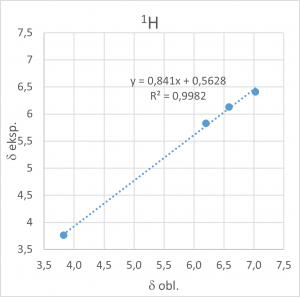 |
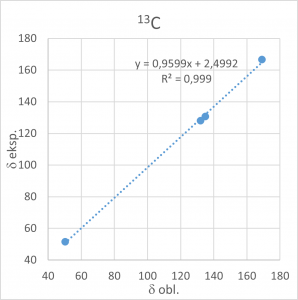 |
Rys. 1.3.2. Wykresy zależności zmierzonych wartości przesunięć chemicznych 1H i 13C od wartości obliczonych dla akrylanu metylu.
Jak widać, w przypadku akrylanu metylu obserwujemy liniową zależność o bardzo wysokim współczynniku korelacji pomiędzy obliczonymi i zmierzonymi wartościami przesunięć chemicznych, zarówno 1H, jak i 13C.
Obliczanie wartości przesunięć chemicznych za pomocą programu Spartan jest dużo prostsze i w pełni zautomatyzowane. Wystarczyć narysować wzór strukturalny cząsteczki związku, którego widmami NMR jesteśmy zainteresowani i wybrać z menu opcję obliczania widma NMR. Program automatycznie analizuje możliwe konformacje cząsteczki i oblicza ich populacje. W kolejnym kroku oblicza dla każdej z nich wartości przesunięć chemicznych, a następnie oblicza średnie ważone na podstawie obliczonych wcześniej populacji konformerów. Dodatkowo wprowadza jeszcze pewne poprawki empiryczne poprawiające dokładność wyników. Zaletą tego podejścia jest łatwość wykonania obliczeń, a wadą – brak możliwości kontroli wyników poszczególnych etapów obliczeń i ich modyfikacji.
Obliczenia wykonane dla octanu winylu zgodnie z oczekiwaniem wskazały na obecność tylko jednego konformeru. Wyniki w oryginalnym formacie są pokazane Tabeli 1.3.3.
Tabela 1.3.3. Wyniki obliczenia przesunięć chemicznych 1H i 13C w cząsteczce octanu winylu za pomocą programu Spartan.
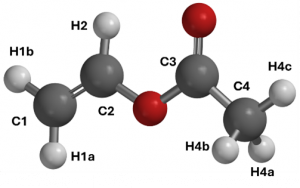
| position | δ C | mult | δ H | splitting (Hz) | |
| 1 | 95.6 | CH2 | 4.77 | dd | (15.3, -1.2) |
| . | 4.51 | dd | (7.2, -1.2) | ||
| 2 | 141.1 | CH | 7.64 | dd | (15.3, 7.2) |
| 3 | 169 | C | |||
| 4 | 20.2 | CH3 | 1.98 | s | |
Dodatkowo program obliczył także wartości stałych sprzężenia H-H. Porównanie obliczonych i zmierzonych wartości przesunięć chemicznych jest pokazane na rys. 1.3.3.
Rys. 1.3.3. Wykresy zależności zmierzonych wartości przesunięć chemicznych 1H i 13C od wartości obliczonych dla octanu winylu za pomocą programu Spartan.
Jeśli porównany wartości przesunięć chemicznych obliczonych za pomocą programów Gaussian i Spartan, to zobaczymy znaczące różnice pomiędzy nimi. Jednak także w przypadku wyników uzyskanych za pomocą Spartana zależność pomiędzy obliczonymi i zmierzonymi wartościami przesunięć chemicznych 1H i 13C jest prostoliniowa z bardzo dobrym współczynnikiem korelacji.
Porównanie zmierzonych i obliczonych stałych sprzężenia także wskazuje na ich dobrą – chociaż nie idealną – zgodność. Pokazane jest to na poniższym rysunku.
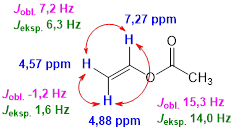
Taka zgodność jest całkowicie wystarczająca dla potwierdzenia poprawności interpretacji widma.
Także w przypadku akrylanu metylu wyniki uzyskane za pomocą programu Spartan są bardzo podobne zarówno do obliczonych przy użyciu Gaussiana, jak i do wyników eksperymentalnych. Spartan zidentyfikował dwa konformery i obliczył różnicę swobodnej energii Gibbsa pomiędzy nimi równą 0,76 kcal/mol, co oznacza, że populacja trwalszego konformeru wynosi 78%, a mniej trwałego – 22%. Jest to wynik niemal identyczny z uzyskanym za pomocą Gaussiana (76% i 24%). Obliczone wartości przesunięć chemicznych i stałych sprzężenia są pokazane w Tabeli 1.3.4.
Tabela 1.3.4. Wyniki obliczenia przesunięć chemicznych 1H i 13C w cząsteczce akrylanu metylu za pomocą programu Spartan.
| position | δ C | mult | δ H | splitting (Hz) | |
| 1 | 133 | CH2 | 5.73 | dd | (10.9, 1.6) |
| . | 6.5 | dd | (18.6, 1.6) | ||
| 2 | 127.8 | CH | 6.22 | dd | (18.6, 10.9) |
| 3 | 167.3 | C | |||
| 6 | 52.1 | CH3 | 3.77 | s | |
Porównanie obliczonych i zmierzonych wartości przesunięć chemicznych jest pokazane na rys. 1.3.4. Tu także zgodność jest bardzo dobra.
Rys. 1.3.4. Wykresy zależności zmierzonych wartości przesunięć chemicznych 1H i 13C od wartości obliczonych dla akrylanu metylu.
Zmierzone i obliczone wartości stałych sprzężenia są pokazane na poniższym rysunku.
Jak widać, dla cząsteczki akrylanu metylu zgodność obliczonych i zmierzonych stałych sprzężenia jest znakomita.
Podsumowując ten podrozdział należy stwierdzić, że do rutynowego stosowania jako narzędzia pomocniczego przy interpretacji widm NMR program Spartan jest dużo wygodniejszy, a wyniki, które daje, są w pełni akceptowalne. Wymaga jednak wykupienia licencji oraz posiadania wydajnego komputera (zalecany jest procesor 16-rdzeniowy) – przynajmniej do obliczeń większych cząsteczek. Program Gaussian jest z kolei dostępny za darmo dla pracowników naukowych i studentów na uczelniach wyższych i w instytutach naukowych za pośrednictwem systemu PLGrid (https://plgrid.pl/). Obliczenia są wykonywane bardzo szybko, ale trzeba nauczyć się tworzenia plików wsadowych do obliczeń oraz odczytywania właściwych danych z pliku wynikowego. Może w tym pomóc wspomniany już w podrozdziale 1.3.4 program GaussView. Można też zakupić licencję na używanie Gaussiana w wersji dla systemu Windows lub Linux na komputerze osobistym, ale trzeba się liczyć ze znacząco dłuższym czasem obliczeń dla cząsteczek większych niż 30 atomów.